在分子生物学与生物信息学分析过程中,序列比对是一项至关重要的工作。无论是探究基因的同源性、分析蛋白质结构的保守区域,还是构建系统发育树,序列比对结果的准确解读与效率控制都直接影响分析结果的可靠性与科研效率。DNAMAN作为一款经典的图形化生物序列处理软件,以其简洁直观的界面和较强的可视化能力,在教学和中小规模科研项目中广泛使用。那么,DNAMAN比对结果怎么看?在执行多重比对时,DNAMAN多重比对速度慢如何优化?本文将从操作方法、图谱解读、性能提升三个方面进行详尽解析,帮助用户更高效地使用DNAMAN进行精准比对分析。
一、DNAMAN比对结果怎么看
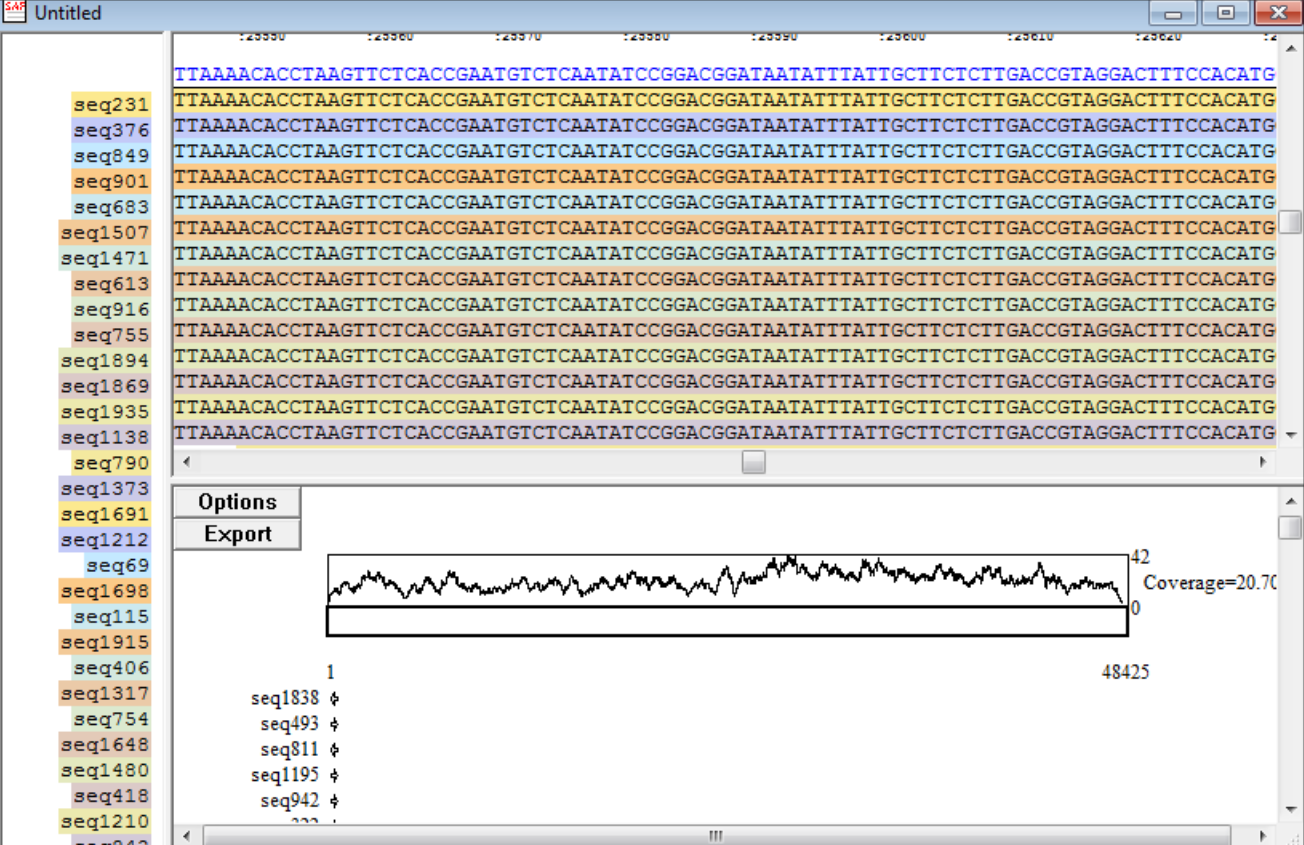
在DNAMAN中执行序列比对主要分为两类:双序列比对(pairwise alignment)和多序列比对(multiple sequence alignment, MSA)。比对完成后,软件会以图形化形式展示比对结果,包括保守区域、高变位点、缺失/插入区域等,便于用户直观观察序列相似性及差异。
1. 多序列比对结果界面介绍
完成ClustalW或其他算法的多序列比对后,DNAMAN会在主窗口展示以下信息:
序列排列区域:所有导入的序列以行的方式排列,每一列表示各序列在该位点的碱基或氨基酸;
保守性标识条(Conservation Bar):在比对图下方显示“*”、“:”、“.”等符号,分别代表完全保守、相似、弱相似;
颜色高亮:根据设置,DNAMAN会使用颜色对不同氨基酸性质进行高亮(如极性、非极性、酸性、碱性),也可以根据保守程度进行着色;
空白与“-”符号:表示比对中存在缺失(gap)或插入;
序列编号与名称:左侧可看到各条序列的名称或编号,便于识别来源。
2. 比对图谱的阅读技巧
识别保守区域:查看哪些位置在所有序列中都一致,是后续设计引物、识别功能位点的重要参考;
观察突变热点:在功能基因中,差异位点可能提示某些突变事件的发生;
Gap区域处理:大量Gap出现可能表示序列拼接或长度不一,需在前处理时标准化长度;
注意氨基酸类别变化:对于蛋白序列,哪怕不是完全一致,也要关注相似氨基酸的替换是否影响蛋白功能。
3. 利用比对结果导出功能
保存比对图谱:点击“File > Export Image”可将比对结果以高清图像格式导出(PNG、TIFF等);
导出比对序列:支持以Clustal格式(.aln)或FASTA格式导出已比对好的序列,便于用于其他分析软件如MEGA、RAxML等;
打印功能:DNAMAN支持分屏打印或分页展示,非常适合做教学材料或实验报告图表。

二、DNAMAN多重比对速度慢如何优化
尽管DNAMAN适用于大多数常规比对任务,但当导入序列数量较多或长度较长时,多重比对的速度可能明显下降,甚至在某些电脑配置下出现卡顿或崩溃的情况。造成比对速度慢的原因主要有两个方面:硬件资源限制与比对算法效率。针对这一问题,用户可以从以下几个方向进行优化。
1. 控制单次比对的序列数量与长度
建议控制在50条序列以内,每条序列长度不超过3000碱基或1000氨基酸;
若必须处理超大数据集,可将其分批比对,再在后期合并分析或拼接系统发育树;
可使用外部工具(如SeqKit、BioPython)先对数据进行长度过滤与冗余序列去重。
2. 关闭实时图谱渲染
在执行比对时,DNAMAN默认开启实时图谱渲染功能,这会占用大量系统资源。可通过如下方法关闭:
在菜单栏选择“Options > Display Settings”;
取消勾选“Live Update of Alignment View”;
等比对完成后再手动刷新视图。
3. 提前统一序列方向和长度
比对算法会尝试自动识别正反链并进行方向匹配,这会增加运算负担。建议在导入前统一序列方向,避免反向互补错误。使用DNAMAN的“Sequence > Complement”或外部脚本可实现。
4. 升级算法与调整参数
DNAMAN默认使用ClustalW或内置算法进行比对,虽然稳定但不如新算法高效。可以:
在比对界面选择“Use Fast Align”(快速比对模式);
调整Gap开销(gap open penalty)与扩展惩罚值(gap extension penalty)来缩短运算时间;
减少高变位点之间的Gap惩罚值,有时可提升效率。
5. 升级硬件配置
若经常进行大规模比对任务,建议提升硬件配置,尤其是:
增加物理内存至16GB以上;
使用SSD提升读写效率;
使用64位系统运行DNAMAN增强版本(如DNAMAN 9+)。
三、如何将DNAMAN比对结果用于后续分析与科研图表
比对结果不仅用于观察序列异同,更可作为后续功能分析、结构预测、系统发育构建的基础。如何从DNAMAN导出的比对结果中获取更多有用信息,是科学研究的加分项。
1. 用于构建系统发育树
将DNAMAN比对结果导出为Clustal格式或FASTA格式;
导入至MEGA、IQ-TREE等软件,选择NJ、ML等算法构建进化树;
保留比对保守区域,有助于提升树结构的准确性;
比对图谱可作为支持性证据附在科研论文中。
2. 用于引物设计与探针开发
从比对图中提取稳定保守区域,用于设计通用引物;
高变区适合设计特异性探针;
结合DNAMAN自带的引物设计功能,可直接从比对结果中调用目标片段,快速生成上下游引物对。
3. 用于结构预测与功能注释
针对蛋白质序列,可识别保守氨基酸位点,预测催化位点、功能域;
对比多个物种的功能基因序列,有助于理解保守结构与变异模式;
将比对结果作为注释参考,匹配KEGG、Pfam、COG等数据库进行功能映射。
4. 论文图表制作
导出高清图像格式的比对结果;
使用图像编辑工具(如Illustrator)微调排版;
配合注释图(如基因结构图、蛋白结构示意图),形成完整的科研图表。

总结
DNAMAN比对结果怎么看 DNAMAN多重比对速度慢如何优化是研究者在日常序列分析中频繁面临的现实问题。DNAMAN凭借直观的可视化界面和稳定的比对功能,在中小规模比对任务中表现良好。通过掌握比对图谱的解读方式、合理控制输入序列数量、关闭不必要的实时渲染功能并进行参数优化,用户可以显著提升比对效率。同时,充分利用DNAMAN生成的比对结果为系统发育树构建、功能区分析、引物设计等后续科研任务提供基础支撑,使其在整个生信分析流程中发挥更大的作用。
