在分子生物学与基因功能研究中,DNAMAN 作为一款专业级序列分析软件,其多序列比对(Multiple Sequence Alignment,MSA)功能被广泛应用于进化分析、保守区域识别及引物设计等领域。然而,许多用户在实际操作中因忽略关键细节导致结果偏差。本文将以“手把手”形式,从数据准备、参数配置到结果优化,深入拆解DNAMAN 多序列比对的全流程技术细节,并提供可落地的进阶方案,助力用户实现精准、高效的序列分析。
一、DNAMAN 多序列比对

1.1数据准备阶段的技术规范
DNAMAN 支持FASTA、GenBank、EMBL等格式,但不同来源的序列需遵循以下预处理原则:
格式统一化:
使用文本编辑器(如Notepad++)确保所有序列标识符格式一致。例如,避免混用“>Gene_1”和“>Sample1_gene”,建议统一为“>SpeciesName_GeneID”(如“>Human_TP53”)。
序列清洁处理:
删除非标准字符:清除序列中的空格、数字或“*”号(终止符),仅保留ATCG或氨基酸代码。
长度均一化:对于长度差异超过30%的序列(如基因片段与全长序列混合),需在DNAMAN 中启用“TrimtoShortest”功能,或手动截取同源区域。
1.2算法选择与参数精细化配置
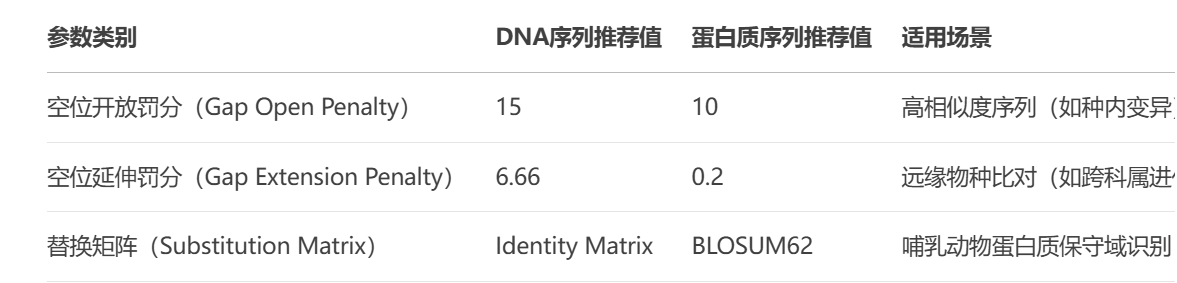
DNAMAN 内置ClustalW、Muscle等算法,用户需根据数据类型调整核心参数:
操作示例:
若比对新冠病毒刺突蛋白(Spike)的变异株序列:
1.选择“Progressive Alignment”算法(适合同源性>80%的序列)。
2.设置空位开放罚分=12、延伸罚分=1.5,降低因插入缺失(Indels)导致的过度分段。
3.使用“BLOSUM80”矩阵,增强对近期进化分支的敏感度。
1.3结果验证与纠错机制
完成比对后,需通过以下方法验证DNAMAN 输出质量:
保守性热图分析:点击“View>Colorby Conservation”,红色高亮区域代表保守位点。若关键功能域(如酶活性位点)未显色,可能需重新调整罚分参数。
人工校对工具:
拖动错位序列区块:按住Ctrl+鼠标左键,可整体移动某条序列的局部区域。
插入强制空位:右键点击目标位点,选择“InsertGap”手动对齐突变簇。
二、DNAMAN 序列对齐操作指南

2.1逐步操作流程图解
步骤1:导入数据并创建项目
1.启动DNAMAN ,点击“File>New>Project”,命名项目(如“2024_HIV_Subtyping”)。
2.选择“ImportSequences”,批量导入待比对文件。关键提示:若导入时提示“FormatError”,需检查文件编码是否为UTF-8(用EmEditor转换)。
步骤2:执行多序列比对
1.进入“Tasks>Alignment>Multiple Alignment”,勾选所有目标序列。
2.点击“Advanced Settings”,按1.2章节配置参数。特别注意:勾选“Use Terminal GapsPenalty”以避免末端冗余空位。
步骤3:结果优化与导出
1.在比对视图中,右键选择“Realign Selected Regions”对高变异区进行局部重对齐。
2.导出前执行“Consensus Threshold”设置:将保守度阈值设为70%,DNAMAN 会自动标记低于此值的位点(便于后续突变分析)。
2.2高频问题解决方案
问题1:比对速度过慢
对策:启用“Low Memory Mode”(适用于>500条序列),或拆分任务为多个子集比对后合并。
问题2:保守区域断裂
对策:在“Alignment Parameters”中降低空位延伸罚分至0.1,并启用“Iterative Refinement”循环3次。
三、DNAMAN 在宏基因组分箱(Binning)中的创新应用
3.1跨工具数据整合方案
DNAMAN 不仅可用于单纯序列比对,还可与宏基因组分析流程联动:
1.Contig聚类预处理:
将Metagenomic组装获得的Contig导入DNAMAN ,执行全基因组比对,识别高相似度Contig集群(阈值设定为ANI>95%),作为分箱的初级标记。
2.标记基因提取:
利用DNAMAN 的“Pattern Search”功能,搜索单拷贝核心基因(如rpoB、gyrB),导出FASTA文件供CheckM评估分箱完整性。
3.2自动化脚本开发示例
通过DNAMAN 内置的Linden Scripting系统,可编写分箱辅助脚本:

DNAMAN 的多序列比对功能,通过精准的参数配置与创新的工作流设计,可满足从基础科研到临床诊断的多样化需求。本文详述的预处理规范、参数优化表及宏基因组分箱案例,不仅解决了用户常见的操作痛点,更为高阶应用提供了技术框架。未来,随着AI算法的集成,DNAMAN 有望实现“一键式”智能比对,进一步降低生物信息学分析门槛,推动精准医学的快速发展。
